A nem megfelelő módon hajtogatott fehérjéket (misfolded, unfolded) közvetlenül a BIP szenzor érzékeli, amely az ER lumenében folyamatosan monitorozza a beérkező fehérjék szerkezeti jellemzőit. Ez a szenzor mechanizmus váltja ki tulajdonképpen a fejezetben eddig tárgyalt UPR-t. Azonban, ha a BIP és / vagy más szenzorok hibásan vagy egyáltalán nem működnek, akkor a fehérje átmehet az ellenőrzőpontokon az esetleges hibák kijavítása nélkül is. Amennyiben az unfolded protein kis mennyiségben van jelen, akkor keletkezik az ún. loss of function fenotípus.
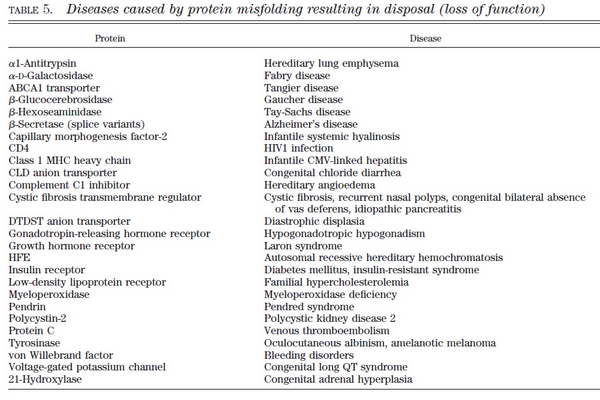
13. ábra Funkcióvesztéses folding – patológia indukált betegségek. Hebert, D. N. and M. Molinari (2007). "In and out of the ER: protein folding, quality control, degradation, and related human diseases." Physiol Rev 87(4): 1377-408.
Abban az esetben, ha az ilyen fehérjék nagy mennyiségben felszaporodnak és akkumulálódnak, gain of toxic fenotípusról beszélünk.
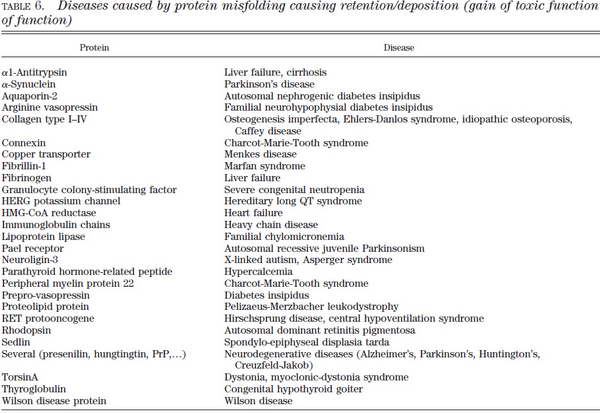
14. ábra Toxikus fehérje akkumuláció indukált betegségek. Hebert, D. N. and M. Molinari (2007). "In and out of the ER: protein folding, quality control, degradation, and related human diseases." Physiol Rev 87(4): 1377-408.
Az ily módon kialakult és krónikusan fennálló ER-stressz pedig a klinikumban is ismert betegségekhez vezethet.
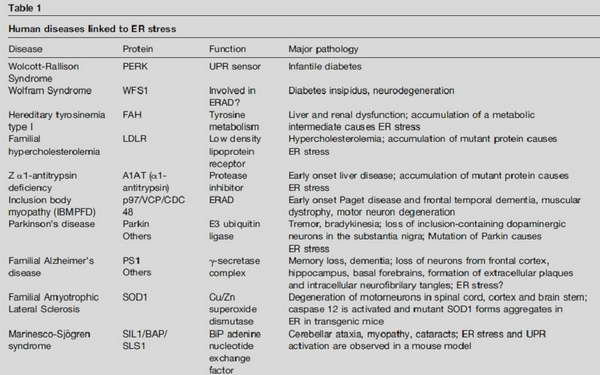
15. ábra ER-stressz kapcsolt humán megbetegedések. Zhao, L. and S. L. Ackerman (2006). "Endoplasmic reticulum stress in health and disease." Curr Opin Cell Biol 18(4): 444-52.
A mellékelt táblázatokból megtudhatjuk, hogy e kórképek mögött olykor csupán egyetlen fehérje folding hibája áll. Legtöbb esetben ezeket a módosulásokat az adott fehérje génjének mutációi okozzák, így hangsúlyoznunk kell a genetikai szűrés fontosságát. Természetesen a táblázatok adatai tájékoztató jellegűek, nem cél minden felsorolt betegség memorizálása, az érdeklődő hallgatók témában történő elmélyedésének kiindulópontjai lehetnek. Az endoplazmatikus retikulum stressz számos jól ismert patológiás állapotban játszhat közvetlenül vagy közvetve fontos szerepet.
Az Anderson-Fabry betegség egy lizoszómális tárolási kórkép, amelyet az alfa-galaktozidáz (lizoszomális hidroláz) hiánya okoz. Funkcionális enzim hiányában felszaporodnak a glikofoszfolipidek a szervezetben (ér endothel, simaizom, bőr, szívizom, cornea, vese, tüdő, bél területén). A változatos lerakódási helyek miatt természetesen a klinikai megjelenése is eltérő. A férfiak homozigóták, esetükben a betegség már serdülőkorban diagnosztizálható. A nők heterozigóták, náluk a betegség enyhébb lefolyású és később is manifesztálódik. Oki kezelése nem ismert. A halált szív, vese, agyi komplikációk okozzák.
A Wilson-kór a réz anyagcsere örökletes rendellenessége, amely egy májspecifikus réz transzporter (cöruloplazmin, WDP, Wilson-diesase protein) mutációja. Változatos, korspecifikus klinikai tünetek jellemzik a szervezet réz felhalmozódását. Májmegnagyobbodás, májspecifikus enzimek extrém magas szintje már gyerekkorban, ascites, nyelőcsővérzés. Központi idegrendszerben gliózis, bazális ganglionok spongiform degenerációja jellemző. Arc és gégeizmok disztóniája miatt disphonia jelentkezhet. A végső stádiumban elbutulás, kóma következik be. Kezelése réz felszívódás gátlók és diéta alkalmazása. Végső esetben máj transzplantáció jöhet szóba. Az enzim külső adagolása átmeneti javulást sem okoz.
Egy másik réztranszporter ATPáz fehérje, az ATP7A mutációja okozza a ritka Menkes szindrómát. Tekintettel arra, hogy a transzporter már a magzati életben sem működik megfelelően, a placentán keresztül nem jutnak réz ionok a központi idegrendszerbe kellő mennyiségben és ez komoly neuronális fejlődészavarhoz vezet. Elektronmikroszkóposan mitokondriális degeneráció bizonyított számos idegsejtféleségben (gerincvelő Clarke-oszlop, neocortex, Purkinje sejt). A betegség rendkívül súlyos, a betegek általában nem érik meg az 5 éves kort sem.
Laron szindróma esetén a növekedési hormon receptor szerkezet károsodik, törpe fenotípus azonosítható. A növekedési hormon koncentrációja normális, azonban a receptor funkcióvesztése miatt nem képes a hormon a sejtben választ indukálni. Magas, cincogó hang, késői serdülés, menarche, normálisnál kisebb nemi szervek, kezek, lábak jellemzik.
A Pelizaeus – Merzbacher betegség egy neurodegeneratív leukodisztrófia, a proteolipid-1 funkcióvesztése. Optikus atrófia, sípoló légvétel, dementia, ataxia, microcephalia, nystagmus szerepel a vezető tünetek közt. Rosszindulatú esetben a születés után közvetlenül manifesztálódik, míg a jóindulatú esetekben akár a felnőttkort is megérheti a beteg.
Tay – Sachs betegség. Ez a kórkép ismét egyetlen fehérje misfoldingjának következménye. A lizoszómális hexózaminidáz–A funkcióvezstése idegrendszeri gangliozid felhalmozódáshoz vezet. A sejtek általában duzzadt fenotípust mutatnak, retina és gerincvelő érintettség leggyakoribb, esetenként a perifériás idegek is károsodhatnak. A halált fertőzés okozza, tüneti kezelése még enzim bevitellel sem megoldott, így prenatalis diagnózisa nagyon fontos.
Charcot – Marie – Tooth szindróma. Szenzo-motoros neuropátia, connexin fehérje hibája miatt. Zsibbadó kéz és láb, elhaló ujjvégek, külső talp élen járás, peroneus atrófia jellemzi.
Vádli elsorvad, lábujjhegyen járás nem kivitelezhető.
Az I. típusú diabetes esetén a pancreas béta sejtjeinek stresszes állapota áll a háttérben. A misfolded inzulin akkumulációja zavarja meg a szekréciót. p58 knockout egér az I. típusú diabetes fenotípust mutatja, amelyből arra következtettek, hogy a hormon termelésének regenerációjáért legalábbis részben a p58 lehet a felelős. A II. típusú diabetes (perifériás inzulin rezisztencia a sejtfelszíni inzulin receptor szupressziójának következménye, amely az emelkedett UPR miatt aktivált proapoptotikus kináz (JNK) aktivitás egyik jele. Tumoros állapotokban, a tumor centrumában a hipoxia aktiválja az UPR-t. Az XBP1 protein expressziója emelkedik, ami a BIP/GRP94 chaperonok expressziós szintjének növekedését vonja maga után. UPR foszforilálja a p53 tumor szupresszort, ami blokkolja a p53 dependens apoptótikus anyagcsere utat, ami következetes tumornövekedéssel jár. XBP1 knockout egerek kisebb méretű tumort növesztettek.
Neurodegenerációs betegségekben a lipid akkumuláció GM1 gangliozidózist eredményez, amelyet az endoplazmatikus retikulumból Ca2+ efflux követ, ez pedig végül stresszt eredményez. A CHOP, JNK és caspase12 szignifikánssan emelkedig apoptótikus neuronban, míg a BIP szintjének emelkedése csak később mérhető. Ez azt jelenti, hogy neuron esetén a misoflding csak másodlagos fontosságú a lipid felhalmozódással szemben. A BIP kofaktorának (SIL1) károsodása Purkinje sejtek esetén apoptózist, következményes ataxiát okoz. A SIL1 mutációja lehet felelős a humán Marinesco-Sjögren szindróma kialakulásának. Ez a betegség egy mentális retardációval, egyensúlyzavarral, izomgyengeséggel, szürke hályoggal járó autoszómális recesszív neurodegeneratív betegség, amely rendkívül ritka, világszerte eddig körülbelül 100 esetet diagnosztizáltak. A fenotípus csak lassan, évek alatt fejlődik ki. Elsősorban a járás megkezdésekor fedezik fel. A hangképzés zavara általában szóformálási nehézségekben mutatkozik meg, a gyermek célirányos mozgása károsodott, ún. céltévesztés lép fel. A végtagok és a törzsizmok innervációjának patológiája mögött nem perifériás, hanem centrális károsodások állnak. A kisagyi vermis atrófiája MRI-vel igazolható. Bizonyos esetekben a szemidegek sorvadása is igazolt. Jelenleg sem megelőzése, sem pedig oki kezelése nem ismert. A szürke hályog tüneteinek enyhítésében kontaktlencse használata, illetve a beszédzavarokon gyógypedagógiai kezelés segíthet. A betegség nem halálos, azonban a felnőtt paciensek is segítségre szorulnak mindennapjaikban, különösen, mert a járásgyengeség a kor előrehaladtával súlyosbodik. Az endoplazmatikus retikulum stressz neurodegenerációban nem primer ok, de a betegség progresszióját jelentősen befolyásolja.
Az UPR-t fénymikroszkóposan is demonstrálhatjuk neopláziás plazmasejtekben, rutin hematoxillin-eozin festést követően.
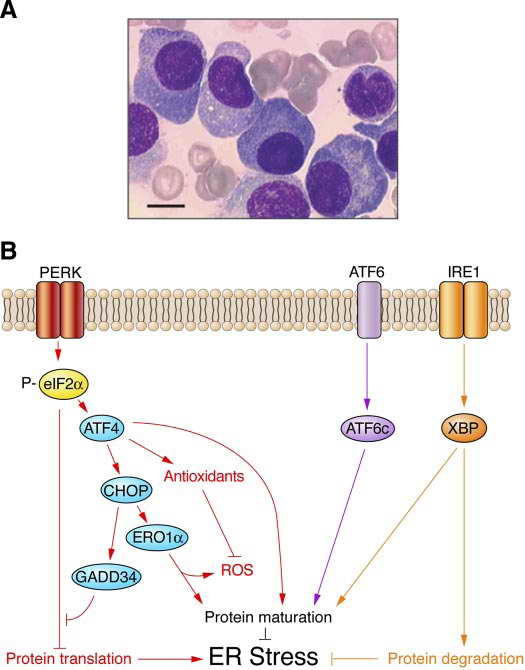
16. ábra. UPR strukturális bizonyítéka plazmasejtekben. Marciniak, S. J. and D. Ron (2006). "Endoplasmic reticulum stress signaling in disease." Physiol Rev 86(4): 1133-49.
Myelómás paciens csontvelőjéből nyert aspirátumon jól megfigyelhetjük a plazmasejtek nukleuszának laterális pozícióba tolódását, amelyet az extrém mértékben felszaporodott endoplazmatikus retikulum okoz. Ez a jelenség a mikroszkópos képen erős bazofiliával jelentkezik. Az ábra továbbiakban sémásan összefoglalja azUPR szignalizációs mechanizmusait, amelyek többek közt szabályozzák a szóban forgó plazmasejtek endoplazmatikus retikulum rendszerének befogadóképességét is. Ilyen esetekben akár ultrastrukturális változásokat is eredményezhet a stresszes állapot, amely plazmasejteknél a sokszor megfigyelt extrém módon kitágult endoplazmatikus retikulum zsákok formájában realizálódik.
Az UPR evolúciósan meglehetősen konzervált folyamat, élesztőben is lezajlik stresszes sejt esetén. A 17. ábrán egy fiziológiás (nem stresszelt) és egy stressz alatt álló sejt endoplazmatikus retikulum szignalizációját tekinthetjük át élesztőben.
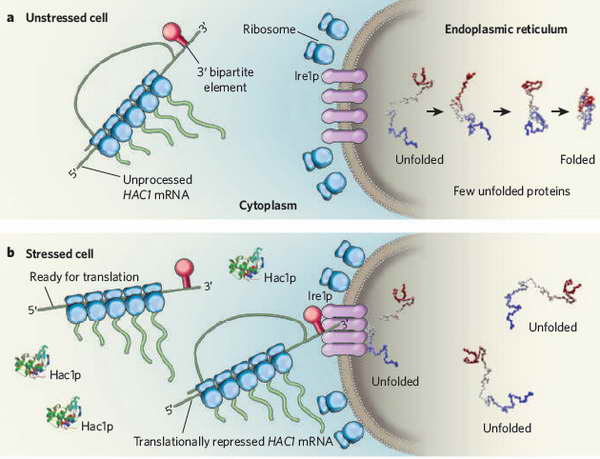
17. ábra. UPR sajátságai élesztőben. Nicchitta, C. V. (2009). "Cell biology: How to combat stress." Nature 457(7230): 668-9.
Kontroll sejt esetén az unfolding szenzor Ire1p inaktív állapotú és a citoplazmatikus HAC1 mRNS (UPR gének expresszióját szabályzó transzkripciós faktor) transzlációja blokkolt. Stressz stimulust követően az Ire1p clusterbe rendeződik és endonukláz aktivitása megemelkedik. A poliriboszómához kapcsolt HAC1 mRNS az aktivált Ire1-hez transzlokálódik és róla megindul az esszenciális stressz faktor Hac1p fehérje szintézise.
Az előző fejezetben az általános sejt stresszre és homeosztatikus folyamatokra fókuszáltunk, majd jelen fejezetben a jóval specifikusabb ER-stressz jelenséget és az ebből következő egyes patológiás állapotokat ismerhettük meg, természetesen a teljesség igénye nélkül. Reméljük, hogy a téma iránt érdeklődő hallgatók számára hasznos kiindulópontként szolgálnak ezek az információk, melyek rávilágítanak arra is, hogy mennyire fontos a sejtszintű homeosztázis fenntartása.