Az autofágia mechanizmusa morfológiai, biokémiai, molekuláris biológiai szempontokból csaknem azonos módon zajlik az élesztőtől a férgeken, a legyeken, a zebrahalon keresztül az egérig és az emberig. Más szavakkal azt mondhatjuk, hogy a folyamat igen erősen konzerválódott a törzsfejlődés során. A benne részt vevő gének, azok szerkezete, funkciója is nagymértékben megörződött. Ezért aztán a kutatók nagyon sok vonatkozásban vizsgálták és vizsgálják az Atg gének ortológjainak működését multicelluláris szervezetekben, így az emlősökben/emberben is. Ezen vizsgálatok igazolták, hogy az Atg gének a törzsfejlődés minden szintjén szükségesek az autofágia folyamatához. Kiesésük, hibájuk gátolja az autofág struktúrák megjelenését, funkcióját. Ez a szervezet működésének súlyos zavaraihoz vezet, sokszor letális.
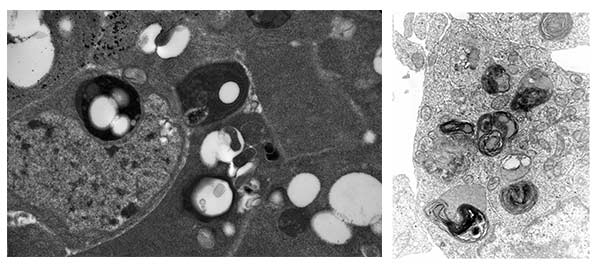
12. ábra. Az autolizoszómák elektronmikroszkópos képe szinte azonos szerkezetet mutat a rovarok zsírtestében (balra) és az emberi agy neuronjaiban (jobbra).
Az Atg gének funkcióinak részletes tanulmányozása számos, fontos eredményre vezetett, amelyek közül néhányat ismertetünk.
13. ábra. Az autofágiát szabályozó génekben okozott mutáció meggátolja a folyamat kialakulását. Felső kép: kontrol, alsó képek: P-elem indukált mutációk.
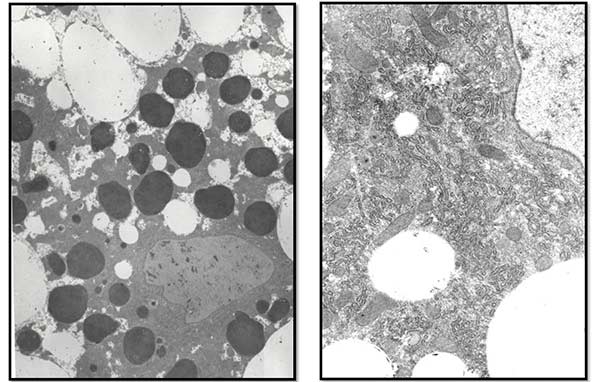
14. ábra. Az elektronmikroszkópos kép még szebben bizonyítja a 13. ábrán bemutatott jelenséget. Balra a kontrol állat, jobbra a mutáns zsírtest sejtjének szerkezete.
15. ábra. Egyes autofágiát szabályozó gének hibája csak a lizoszómák keletkezését zavarja meg és ilyenkor csak autofagoszómák tömege keletkezik a hibás sejtekben (bal oldali kép). Más mutációk miatt éppen az autofagoszómák kialakulása gátolt. Ilyenkor extrém mennyiségű lizoszóma halmozódik fel a sejtekben, de nincsenek autolizoszómák (jobb oldali ábra).
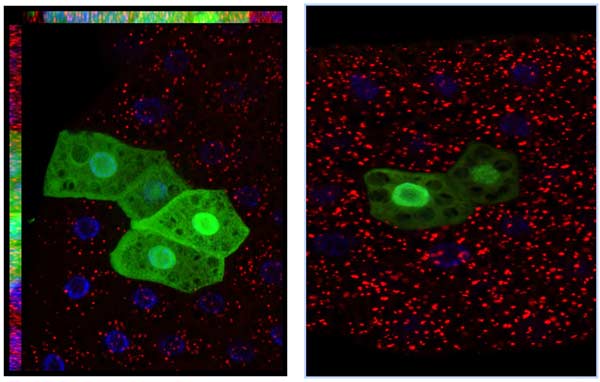
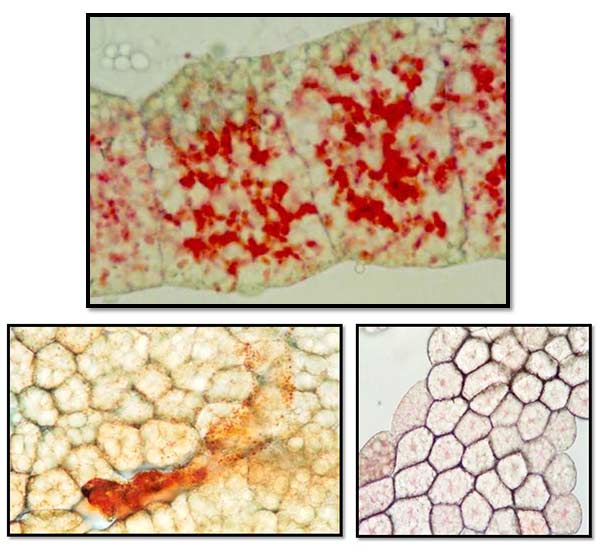
16. ábra. Az autofágia gátlása RNS-interferencia segítségével. A zsírtestben olyan szomatikus klónokat indukáltunk (zöld, GFP-t kifejező klónsejteksejtek), amelyekben az RNS interferencia érvényesült, míg a környező sejtek kontrolként szolgálnak. Jól látható, hogy az autolizoszómák (piros pontok, Atg8 jelölés) csak a kontrol sejtekben jelennek meg, ahol az RNS interferencia gátolta az autofágiát ott nincsenek pirosan jelölődő struktúrák.
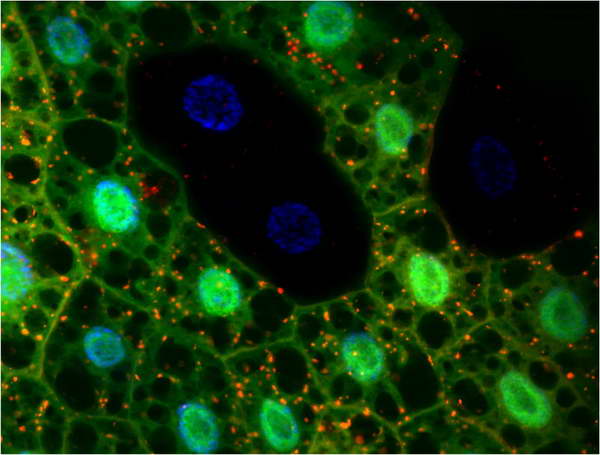
17. ábra. A fenti kísérlet egy olyan rendszerben, ahol a kontrol sejtek fejeznek ki GFP-t, az RNS-interferenciás kontrukciót hordozók nem. Jól látszik, hogy a lizoszómákat festő „Lysotracker” csak a kontrol sejtekben mutatja ki az autolizoszómák jelenlétét.
Drosophilában az Atg gének hibája/hiánya általában gátolja az autofágiát, ami nélkül a lárvális szövetek (pl. lárvális zsírtest, középbél, nyálmirigy) nem bomlanak le a bábozódás előtt és alatt. Ennek következtében az állat elpusztul késői lárva, vagy korai báb állapotban. A rovarok metamorfózisa során extrém magas az autofág aktivitás a lárvális szövetekben. Az erősen felfokozott, túlműködő autofágia a programozott sejthalál egyik formája lehet. Az autofág sejthalál akkor következik be, amikor a fejlődési program (pl. a metamorfózis) vagy a felnőttkori homeosztatikus folyamatok (pl. az emlőmirigy tejelválasztás utáni visszafejlődése, vagy a kasztráció után bekövetkező prosztata és ondóhólyag visszafejlődés) sok sejt egyidejű és együttes eliminációját igényli.
Az Atg7 gén mutációja az autofágia, valamint a DNS fragmentációjának gátlásához vezet. Ezzel ellentétben, az imaginális agyban DNS fragmentálódás és neuronális sejthalál következik be Atg7 mutáns állatokban, vagyis a normális működés során az autofágia (az ún. housekeeping autofágia) megléte a neuronok egészséges működésének fenntartásáért felelős és megakadályozza az idegsejtek pusztulását.
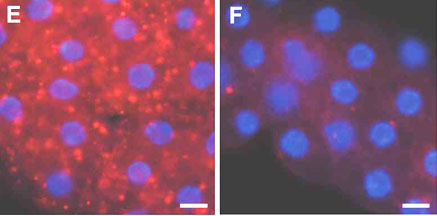
18. ábra. Az Atg7 mutációja meggátolja a zsírtestben az autofág folyamatok beindulását.
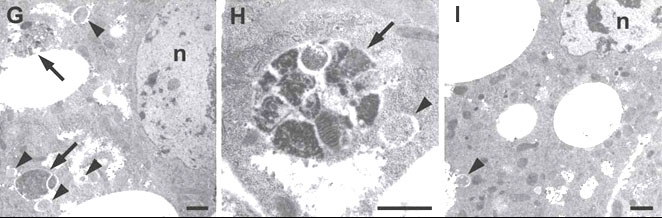
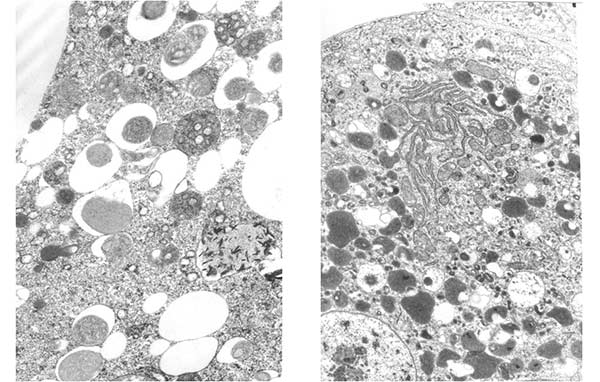
19. ábra. Az Atg7 mutánsok zsirtest sejtjeiben elektronmikroszópos szinten is hiányzik az autofág működés valamennyi jele. G: kontrol sejt, H: autolizoszóma egy kontrol sejtben, I: a mutáns zsírtest sejt szerkezete.
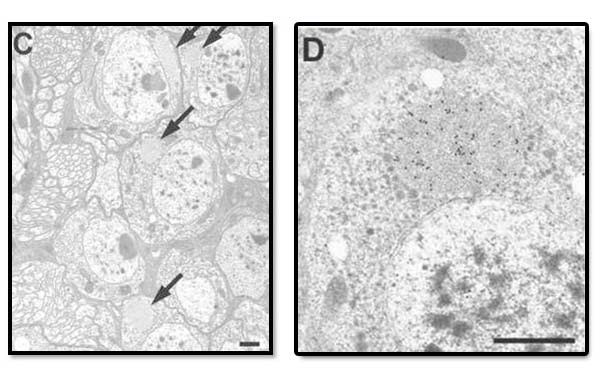
20. ábra. Az autofágia gátlása következtében a legyek neuronjaiban fehérjeaggregátumok (inclusion bodies) szaporodnak fel (C). Ezek ubiquitint tartalmaznak, amint az jól látszik a D képen, ahol az aggregátumokon erős aranyjelölés figyelhető meg.
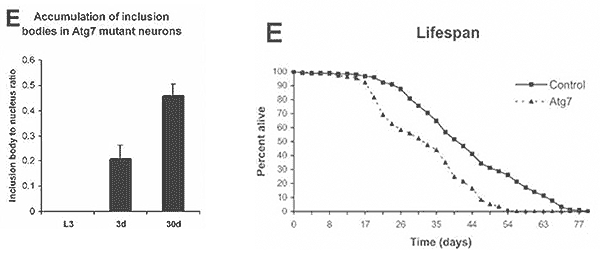
21. ábra. A fehérjeaggregátumok felszaporodása grafikusan (balra) és a legyek élethosszának rövidülése az Atg7 mutációjának következtében (jobbra).
Az Atg5 gén szerepe létfontosságú az emlősök embriogenezisének ún. preimplantációs időszakában. Az Atg5 kiütése (knock out, KO) az egér petesejtben letalitás hatású, az embriók elpusztulnak a 4-8 sejtes állapotban. Az embrionális élet ezen korai szakaszában az autofágia működése feltétlenül kell a fehérjeszintézishez szükséges, magas, citoplazmatikus aminosav koncentráció fenntartásához, valamint a feleslegessé váló fehérjék és organellumok lebontásához.
A születés utáni, ún. neonatális időszakban az autofágia szintje jelentősen megemelkedik valamennyi szövetben, ugyanis a placentán keresztüli tápanyagellátás megszűnése egy rövid ideig tartó „éhezési” periódust okoz, továbbá egy kifejezett „hősokk” (lehülés) is éri az éppen megszületett kis állatot. Ezt az időszakot autofágia hiányában legtöbbször nem éli túl a szervezet, ugyanis az aminosav szint erősen lecsökken a vérben (és a sejtekben is). Ehhez hozzájárulhat az, hogy a neuronokban az autofágia kimaradása neuronvesztéshez vezet, aminek következtében az utód nem, vagy csak kevéssé tud szopni és ez tovább súlyosbítja az egyébként is fennálló aminosavhiányt.
A vörösvértestek differenciációja során normális körülmények között eltűnik sejtekből a sejtmag és számos más organellum, feleslegessé váló fehérje is. Mindezek elmaradnak az autofág génekben hibás egerekben, aminek következtében a fejlődő szervezetben nem tudnak kialakulni az ép, működőképes vérsejtek és az állat elpusztul. A reticulocytákban a mitokondriumok lebomlása, eliminációja elsősorban az autofágiától függ. Az Atg7-/- egerek erythrocytáiban felhalmozódnak a felesleges, vagy hibás mitokondriumok, ami korai pusztulásukhoz vezet.
Az Atg gének kiütésének hatása különböző morfogenetikai elváltozásokat is okozhat az ontogenezis során. Pl. nem záródik a proamnioticus csatorna, hibásan fejlődik a szív és a máj, vagy pl. az idegszövet extrém módon hiperproliferál.
A humán krónikus neurodegeneratív kórképekben (pl. Alzheimer-, Huntington-, Parkinson-kór) egyes neuronpopulációk az évek során fokozatosan degenerálódnak, majd elpusztulnak. A mutáns vagy toxikus fehérjék felhalmozódása főszerepet játszik ezen betegségekben. E fehérjék degradációjában normális körülmények között jelentős szerepet játszhat az autofágia, mint a sejt egyik fő lebontó apparátusa. Ennek megfelelően sokan az autofág struktúrák felhalmozódását figyelték meg a betegek idegsejtjeiben. Számos olyan eredmény született, amely az autofágia citoprotektív funkcióját igazolta a neuronvesztéses kórképekben.
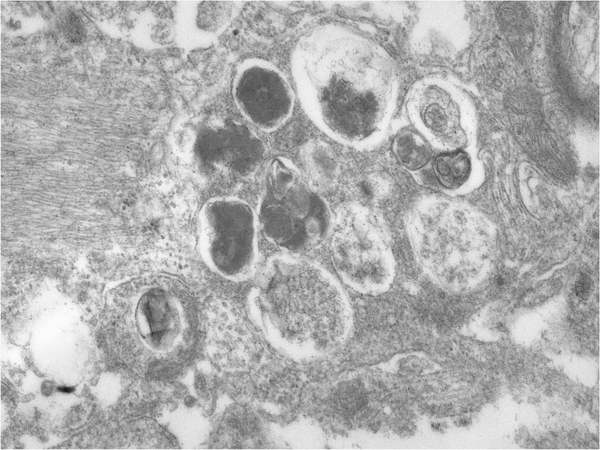
22. ábra. Intenzív autofágia egy pusztuló idegsejtben
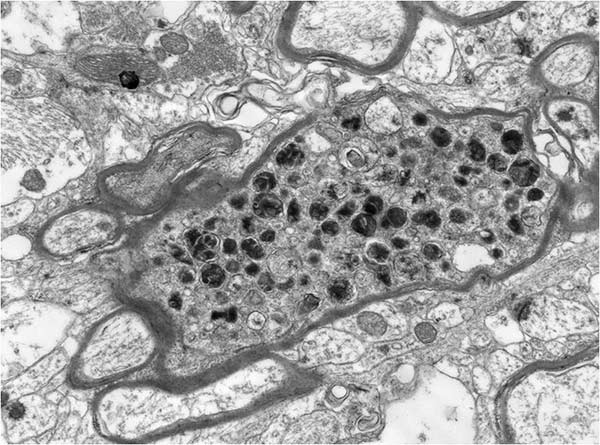
23. ábra. Egy Alzheimer kórban szenvedő beteg pusztuló neuronjának ún. disztrófiás neuritje
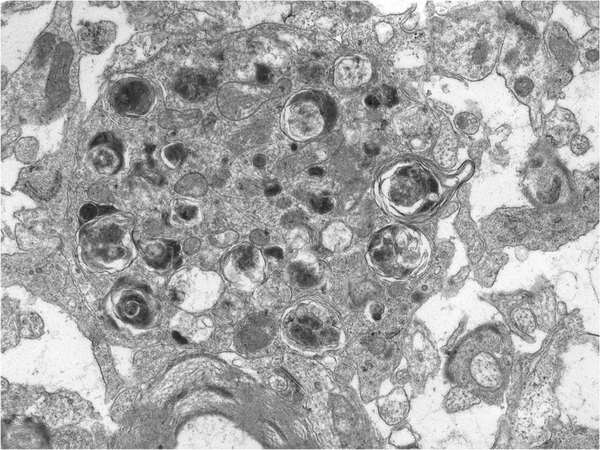
24. ábra. Az ilyen extrém módon felerősödött autofágia minden bizonnyal az idegsejt pusztulásához vezet
Mindezeket alapul véve megpróbálták az autofágia serkentésével mérsékelni a neuronok pusztulását olyan egerekben, amelyek a kóros fehérjéket expresszálták idegsejtjeikben. Rapamycin kezelést alkalmaztak, ami a TORC1 gátlásán keresztül autofágiát indukál a sejtekben. Ez a kezelés felgyorsította a motoros neuronok degenerációját és általában az autofágia indukciója az idegsejtek gyors pusztulásához vezetett. Tehát az várt eredménynek éppen az ellenkezőjét tapasztalták, a felerősített autofágia nem megvédte az idegsejteket, hanem gyors a pusztulásukhoz vezetett.
Az I. típusú diabetes mellitus az a metabolikus rendellenesség, amelyet folyamatosan magas vércukorszint jellemez, mivel az az inzulin termelése és szekréciója csak alacsony szinten zajlik a szervezetben. Ez legtöbbször annak a következménye, hogy a hasnyálmirigy Langerhans-szigeteinek β-sejtjei kevesen vannak és kevés inzulint termelnek. Számos megfigyelés igazolja, hogy diabetesben a β-sejtek száma erősen csökken, ezért a szervezet nem tud a növekvő vércukor szintre inzulin szekrécióval reagálni. A β-sejtek pusztulásában szerepet játszó autofágia megítélése szintén ellentmondásos, ahogy azt a neuronpusztulások esetében már láttuk. Sok eredmény szól amellett, hogy az autofágia megvédi a β-sejteket, más kísérletek eredményei viszont azt bizonyítják, hogy a pusztulásuk a felerősödő autofágia következménye.
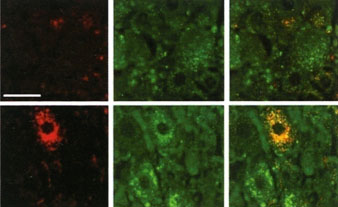
25. ábra. Egy I. típusú diabetes mellitusban szenvedő páciens β-sejtjeiben intenzív autofágiát mutat az Atg6 és az Atg8 géntermékek jelőlése. Balra Atg8, középen Atg6, jobbra a két, egymásra fedő kép.
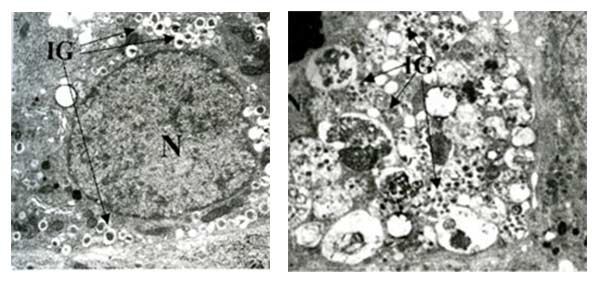
26. ábra. A magas vércukor szint a β-sejtekben először inzulin túltermelést indukál, ami rövidesen ER-stresszt okoz (bal oldali kép, IG: inzulin tartalmú vezikulumok). Az ER-stressz ezután intenzív autofágiát serkent, amint az a jobb oldali képen látszik.
A fentiekben leírtak alapján tehát elmondhatjuk, hogy ma már tudjuk, hogy az autofágia számos betegségünk hátterében álló, különösen fontos sejttani mechnizmus, aminek szerepe van számos patológiás elváltozás kialakulásában is. Arra vonatkozóan is számos ismeret halmozódott fel az utóbbi néhány évben, hogy az autofágia mögött milyen molekuláris mechanizmusok húzónak meg. Ennek kapcsán jó néhány olyan molekula került az érdeklődés középpontjába, amelyek a gyógyszertervezés alapjait képezhetik.
Ugyanakkor nem tudjuk ma sem pontosan, hogy egy adott kórképet az autofágia hiánya, vagy éppen túlműködése okoz, vagy az adott páciens esetében az autofágia citoprotektív, vagy ellenkezőleg, sejtpusztító hatását kellene növelni, vagy csökkenteni a gyógyulás érdekében? Ezért jelenleg a területen inkább az egyéni terápiás kezelések kidolgozásához szükséges diagnosztikumok fejlesztése folyik, mint az autofágiára általában ható gyógyszerek tervezése, vagy termelése. Bizonyosnak látszik azonban, hogy nincs messze az az idő, amikor elegendő adat áll majd rendelkezésre az autofágiát befolyásoló terápiás eljárások kidolgozásához is.