Amint azt a bevezetőben már említettük, nem létezik egyetlen, a sejttérfogatra vonatkozó normálérték (volume set point), hanem a sejt mindig az aktuális szituációnak megfelelően módosítja a térfogatot a korrekciós mechanizmusok (RVI, RVD, organikus ozmolitok) segítségével. A kialakult térfogat változás azonban, nem csupán egy sejtválasz, hanem számos fiziológiás reakció aktivátora, befolyásoló tényezője is. A teljesség igénye nélkül a sejttérfogat változás befolyásolni képes a proliferációt, migrációt, sejthalált, transz epitheliális transzportot, a hormon és transzmitter kibocsátást, illetve antigén antitest reakciót is.
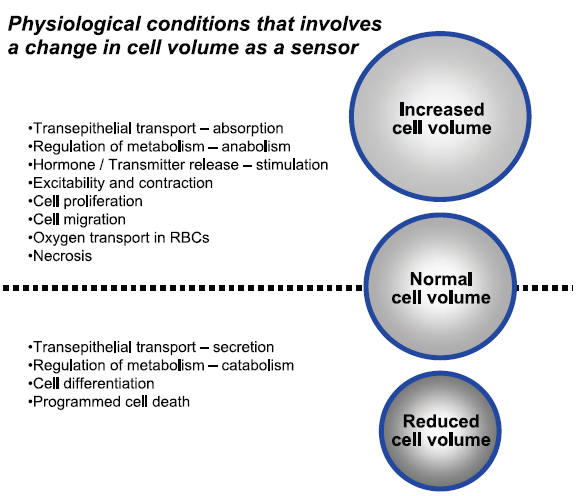
10. ábra. Fiziológiás állapotok és sejttérfogat változás kapcsolata. Hoffmann, E. K., I. H. Lambert, et al. (2009). "Physiology of cell volume regulation in vertebrates." Physiol Rev 89(1): 193-277.
A transz epitheliális iontranszport folyamatos kihívás a sejttérfogat állandóságának fenntartása szempontjából. A 11. ábra három sejtféleség példáján keresztül mutatja be a sejtfunkció és térfogat-szenzáció kapcsolatát. Vékonybél, epehólyag, vese proximális tubulus sejtjei luminális felszínükön a glükózt és aminosavakat (AA) Na+-kapcsolt transzportfolyamatok segítségével veszik fel. A nagy mennyiségű Na+ sejtduzzadást okoz, amely elkerülésére a bazális membrán K+ csatornái nyílnak.
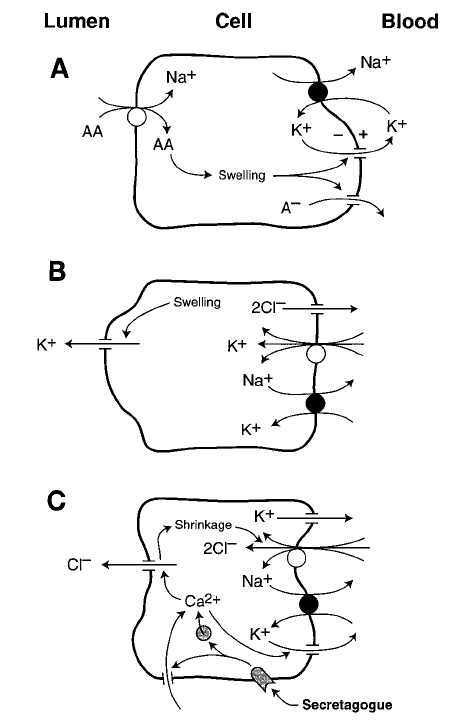
11. ábra. Sejttérfogat és membrántranszport folyamatok kapcsolata. Lang, F., G. L. Busch, et al. (1998). "Functional significance of cell volume regulatory mechanisms." Physiol Rev 78(1): 247-306.
A belső fül vestibuláris sejtjeinek apikális K+ csatornái sejtduzzadásra, míg a bazolaterális NKCC1 sejtzsugorodásra aktiválódnak. A sejttérfogat tulajdonképpen funkciójukban szorosan összekapcsolja az apikális és a bazolaterális membránt, hiszen a túlzott NKCC1 funkció sejtduzzadáshoz vezet, így aktiválja az apikális K+ csatornákat. Ennek működése következetes sejtzsugorodást okoz, ezáltal bekapcsolja az NKCC1-et. A belső fül fontos mechanoszenzor sejtjeinél állandóan fennáll az ionikus homeosztázis változás lehetősége, ezért a fent vázolt módszer folyamatosan monitorozza a sejt térfogatát.
A különböző epitheliumsejtek szekréciós aktivitás alatti K+ és Cl- csatorna nyitása sejtzsugorodáshoz vezet a KCl vesztés miatt. Ez a zsugorodás aktiválja a térfogat érzékeny Na+/H+ cserélő illetve NKCC1 mechanizmusokat, amelyek regenerálják a térfogatot. Az előbb vázolt három sejt tökéletes példája a sejt két ellentétes pólusa közt szinte észrevétlenül megvalósuló információcserének. A sejttérfogat változásra a celluláris pH is érzékeny. A vezikuláris pH lúgosodása pedig alapvetően befolyásolja a szekretoros proteinek vezikuláris szállítását és a membránba történő inszercióját is. A sejttérfogat változása bizonyítottan módosítja a zonulae occludentes permeabilitását, így a paracelluláris transzportot.
A sejtduzzadáskor fellépő membrán tenzió emelkedés kiváltja a plazmamembrán és az endocitótikus vezikulák fúzióját, ami a vezikula tartalmának kiürítéséhez vezet. Ehhez még Ca2+ és intakt aktin citoszkeleton szükséges. Szekretoros vezikulák esetében elmondható, hogy a sejtduzzadás emeli a hormonfelszabadítás és szekréció frekvenciáját.
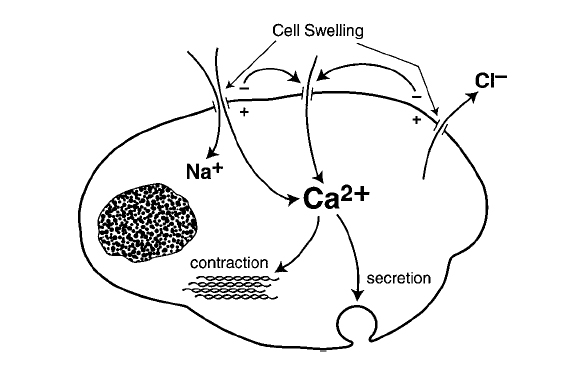
12. ábra A szekréciós tevékenység és a sejttérfogat változás. Lang, F., G. L. Busch, et al. (1998). "Functional significance of cell volume regulatory mechanisms." Physiol Rev 78(1): 247-306
Az inzulin, a prolactin, az aldoszteron, a luteinizáló hormon és a thyrotropin esetében már bizonyították az egyenes arányosságot a sejt duzzadása és a szekréció frekvenciája közt. A folyamat korrelált az intracelluláris Ca2+ koncentráció emelkedésével. Ozmotikus sejtzsugorodás gátolta a prolactin szekréciót (a Ca2+ influx gátlása által). A hiperozmolaritás stimulálja a bazofil granulociták histamin és a C típusú primer szenzoros neuronok Substance P szekrécióját.
A sejtek migrációja a citoszkeleton átrendeződését igénylő komplex folyamat, amelyet polarizáltan szervezett, sorozatos sejt térfogat változások kísérnek.
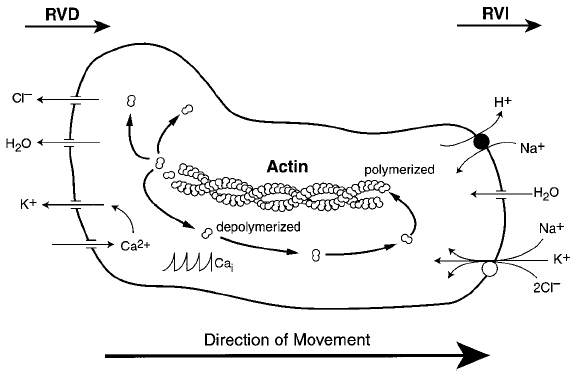
13. ábra. Sejttérfogat változás és migráció. Lang, F., G. L. Busch, et al. (1998). "Functional significance of cell volume regulatory mechanisms." Physiol Rev 78(1): 247-306
A sejt haladási irány felé néző pólusán (leading edge) emelkedett az actin polimerizáció, míg az ellentétes póluson aktin depolimerizációt figyelhetünk meg. A depolimerizációt a Ca2+ vezérelt gelsolin végzi. Az aktin fragmentumok gyors transzporttal a leading edge-hez jutnak, ahol újrahasznosulnak az aktin elongációban. A meglévő aktin hálózathoz új elemek hozzáadását a sejtmembrán protrúziója stimulálja, amely lokális ozmotikus duzzadás eredménye lehet. Az intracelluláris Ca2+ aktivitás (Ca2+i) oszcillációja a sejt haladással ellentétes pólusán Ca2+ szenzitív K+ csatornákat aktivál, amely RVD-hez vezet. Az emelkedett Ca2+ szint gyorsítja a gelsolin működését, következésképp az aktin depolimerizáció is emelkedik. A leading edge területén a NHE1 és a NKCC1 RVI indukciót okoz. A hipertóniás stressz gátolja a neutrofil granulociták migrációs képességét, ugyanakkor a sejt duzzadás szükségesnek bizonyult a folyamathoz. A duzzadás-indukált Cl- és a volume regulált anion csatornák (VRAC) aktivitása szintén a migráció elősegítő faktorainak tekinthető. A transient receptor potencial (TRP) ioncsatorna család két tagja TRPV1 és TRPV4 bizonyítottan fontos mediátor szerepet játszik az ozmoszenzációban és következésképp a sejtmigrációra is hatást gyakorol. A víztranszportra specializálódott Aquaporinok (AQP), azzal hogy facilitálják az ozmotikus vízbeáramlást a membrán protrúziók területére, a sejtek előrehaladását mediálják.
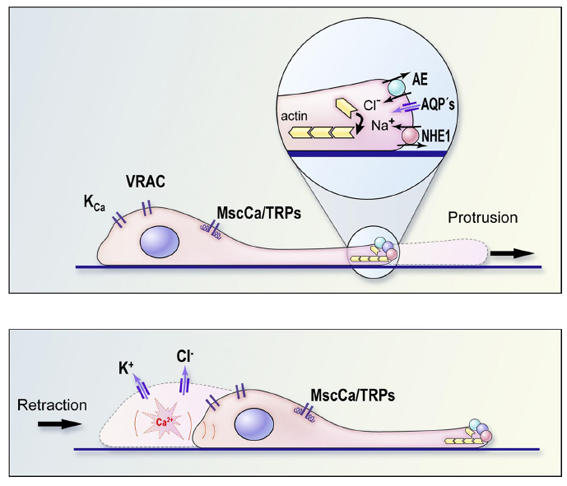
14. ábra. Iontranszport és sejt migráció. Hoffmann, E. K., I. H. Lambert, et al. (2009). "Physiology of cell volume regulation in vertebrates." Physiol Rev 89(1): 193-277.
A programozott sejthalál (PCD) egyik morfológiai jellemzője a jelentős sejtzsugorodás, amelyet apoptótikus térfogatvesztésnek (apoptotic volume decrease, AVD) neveznek. Az AVD egy izozmotikus sejtzsugorodás és általában a PCD bekövetkezését megelőző jelenség. Meg kell jegyeznünk, hogy az AVD alatt a sejttérfogat korrekciós folyamatok (RVI) gátolt állapotban vannak. A sejtzsugorodás által kiváltott sejthalál komplex szignalizációs folyamatait a 15. ábra foglalja össze.
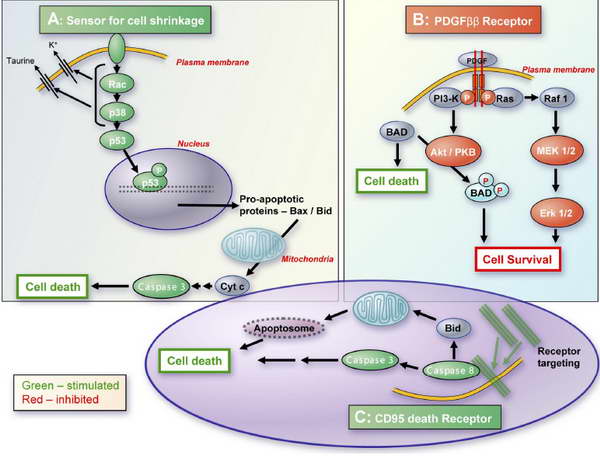
15. ábra. Sejtzsugorodás és programozott sejthalál. Hoffmann, E. K., I. H. Lambert, et al. (2009). "Physiology of cell volume regulation in vertebrates." Physiol Rev 89(1): 193-277.
A zöld szín a kísérletesen igazolt stimulátoros, míg a piros az inhibitoros útvonalakat jelzi. A fejezetben említett térfogat szenzorok valamelyike érzékeli a sejtzsugorodást és aktiválja a monomer GTP kötő proteint (Rac) és p38 MAPkinázt, ezt követi a p53 foszforilációja és nukleáris transzlokációja. A p53 apoptózisban betöltött központi szerepét már 1993-ben leírták, több pro-apoptotikus gén transzkripcióját aktiválja, amelyek azután az intrinsic, mitokondriális (pl. BCL-2 család) és az extrinsic, receptor mediált apoptótikus útvonal (pl. Fas) kulcsenzimei lesznek. A p53 a mitokondriális citokróm-c útvonalon caspase-3 aktivációt eredményez. A Na+/K+ pumpa gátlása AVD alatt K+ efluxot indukál. A K+ csökkenés mellett ilyenkor az organikus ozmolitokhoz tartozó taurin koncentráció csökkenése is megfigyelhető. A sejtzsugorodás gátolja a PDGF receptor mediált szignalizációt. Az így lecsökkent protein kináz B (PKB) működés révén a Bcl-2 család pro apoptótikus proteinjének (BAD) foszforilációja zavart szenved és emelkedik a BAD-mediált sejthalál valószínűsége. Ezzel párhuzamosan a PDGF útvonal gátlása csökkenti a mitogén aktivált protein kináz kináz (MEK, MAPKK) és az extracelluláris szignál regulált kináz (ERK) szignalizációt, ami csökkenti a sejt túlélését. A CD95 sejthalál receptor fiziológiás körülméynek közt intracelluláris lokalizációt mutat. Hiperozmotikus stresszre azonban a plazmamembránba transzlokálódik és caspase 3 és 8 aktivációt indukál. Az említetteken kívül természetesen számos más sejtfunkció esetén figyelhetünk meg sejttérfogat változást és/vagy a korrekciós mechanizmusok aktiválását. A fagocitózis nyilvánvalóan növeli a sejttérfogatot, a Kuppfer sejtek betain tartalmukat felszabadítják a fagocitózis alatt. Az erithrocyták és thrombocyták extrém mértékű morfológiai változásait természetesen jelentős térfogat korrekció kíséri.
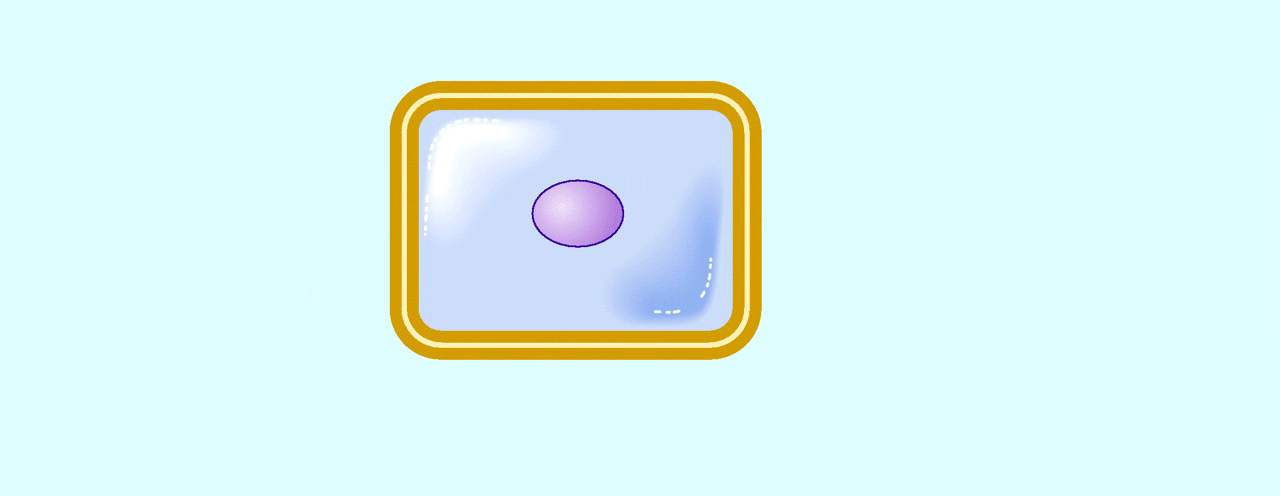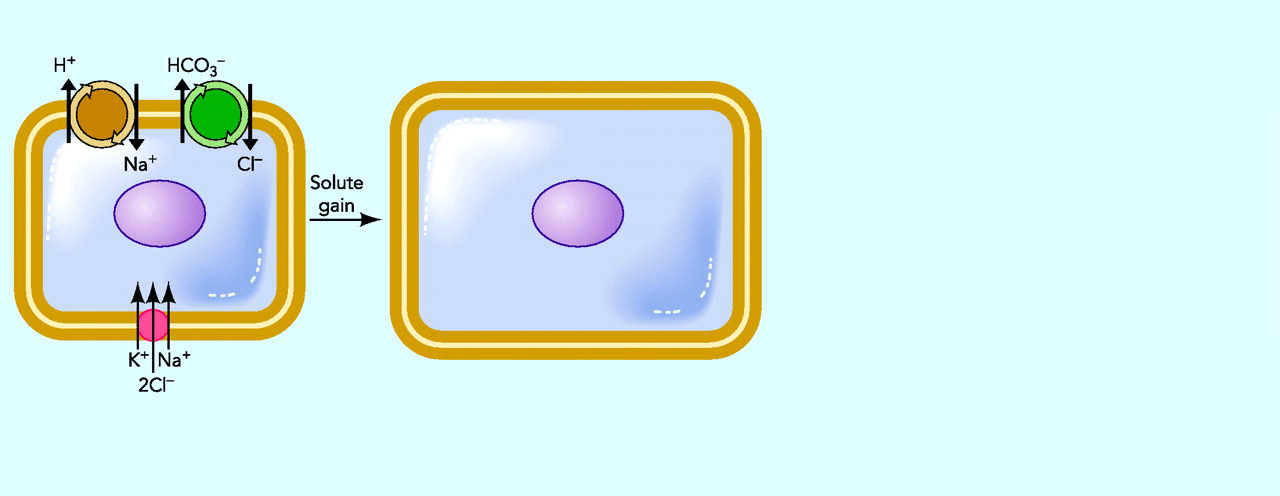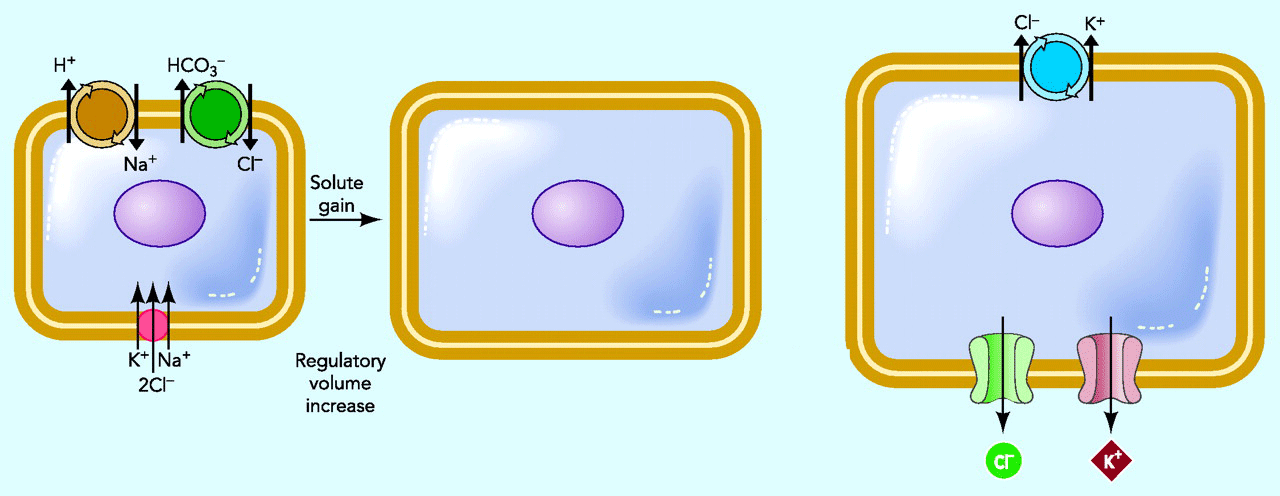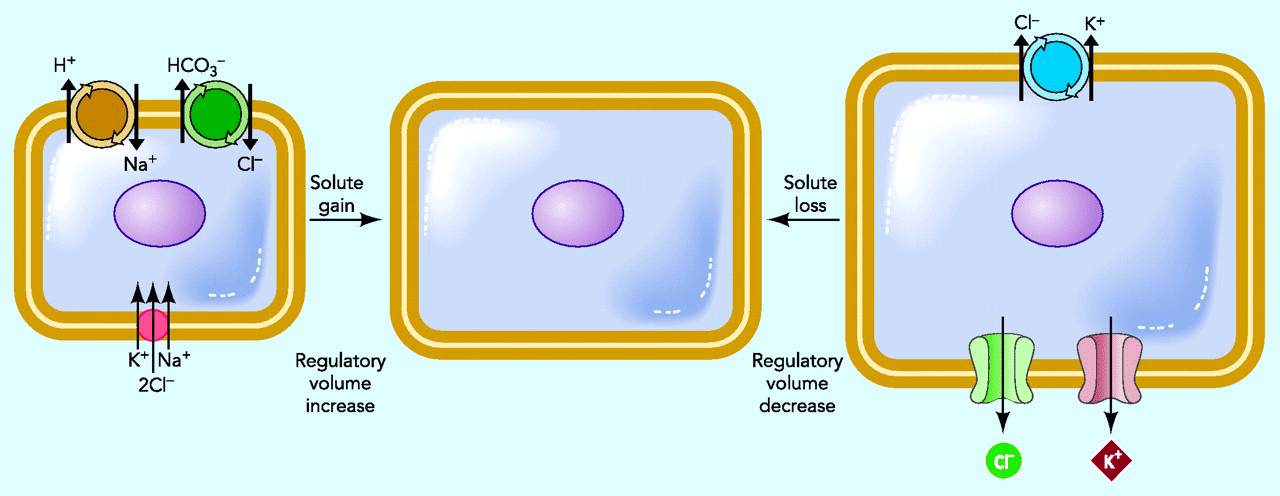 |
16. ábra